class: center, middle, inverse, title-slide # Statistical analysis of a ‘simple’ progeny test ### Facundo Muñoz<br/><a href="mailto:facundo.munoz@cirad.fr">facundo.munoz@cirad.fr</a><br/><i class="fab fa-twitter"></i> <i class="fab fa-github"></i> famuvie ### Orléans, Sep. 18, 2018 --- background-image: url(img/progeny_test.jpg) background-size: cover --- class: middle, inverse # Excercise 1 ## Fit a progeny model to the globulus dataset with __breedR__ Use variables `phe_X` as response and `mum` as grouping variable from the `globulus` dataset (included with `breedR`) .left[ ```r ## Template fm0 <- remlf90( fixed = y ~ 1, # response + fixed effects random = ~ g, # random effects data = D) # dataset ``` ] --- class: inverse, center, middle # Progeny Tests --- ### Elements of a dataset - __response__ variable `\(y\)` - trait of interest, only 1 ('simple' remember?) - __ancestor__ `\(g\)` - or genotype, genetic group, family, provenance, or any __grouping__ variable. --- ### Globulus dataset ```r knitr::kable(head(globulus), format = "html") ``` <table> <thead> <tr> <th style="text-align:right;"> self </th> <th style="text-align:right;"> dad </th> <th style="text-align:right;"> mum </th> <th style="text-align:left;"> gen </th> <th style="text-align:left;"> gg </th> <th style="text-align:left;"> bl </th> <th style="text-align:right;"> phe_X </th> <th style="text-align:right;"> x </th> <th style="text-align:right;"> y </th> </tr> </thead> <tbody> <tr> <td style="text-align:right;"> 69 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 64 </td> <td style="text-align:left;"> 1 </td> <td style="text-align:left;"> 14 </td> <td style="text-align:left;"> 13 </td> <td style="text-align:right;"> 15.756 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 0 </td> </tr> <tr> <td style="text-align:right;"> 70 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 41 </td> <td style="text-align:left;"> 1 </td> <td style="text-align:left;"> 4 </td> <td style="text-align:left;"> 13 </td> <td style="text-align:right;"> 11.141 </td> <td style="text-align:right;"> 3 </td> <td style="text-align:right;"> 0 </td> </tr> <tr> <td style="text-align:right;"> 71 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 56 </td> <td style="text-align:left;"> 1 </td> <td style="text-align:left;"> 14 </td> <td style="text-align:left;"> 13 </td> <td style="text-align:right;"> 19.258 </td> <td style="text-align:right;"> 6 </td> <td style="text-align:right;"> 0 </td> </tr> <tr> <td style="text-align:right;"> 72 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 55 </td> <td style="text-align:left;"> 1 </td> <td style="text-align:left;"> 14 </td> <td style="text-align:left;"> 13 </td> <td style="text-align:right;"> 4.775 </td> <td style="text-align:right;"> 9 </td> <td style="text-align:right;"> 0 </td> </tr> <tr> <td style="text-align:right;"> 73 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 22 </td> <td style="text-align:left;"> 1 </td> <td style="text-align:left;"> 8 </td> <td style="text-align:left;"> 13 </td> <td style="text-align:right;"> 19.099 </td> <td style="text-align:right;"> 12 </td> <td style="text-align:right;"> 0 </td> </tr> <tr> <td style="text-align:right;"> 74 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 50 </td> <td style="text-align:left;"> 1 </td> <td style="text-align:left;"> 14 </td> <td style="text-align:left;"> 13 </td> <td style="text-align:right;"> 19.258 </td> <td style="text-align:right;"> 15 </td> <td style="text-align:right;"> 0 </td> </tr> </tbody> </table> --- ### Statistical model We __assume__ that the observed value for an individual from group `\(g\)` is <!-- $$ --> <!-- \begin{aligned} --> <!-- y_g &\sim \mathcal{N}(\mu_g, \sigma^2) \\ --> <!-- \mu_g &\sim \mathcal{N}(\mu_0, \sigma_g^2) --> <!-- \end{aligned} --> <!-- $$ --> <!-- Use a traditional "mixed effects" formulation which will be more --> <!-- familiar to participants --> $$ y_g \sim \mu_0 + \mu_g + \varepsilon$$ with `\(\mu_g \sim \mathcal N(0, \sigma_g^2)\)` and `\(\varepsilon \sim \mathcal N(0, \sigma^2)\)` .center[ 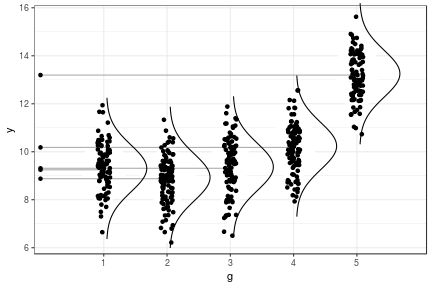<!-- --> ] --- class: middle, inverse # Excercise 2 ## Include further effects 1. Extend `fm0` by including a __fixed effect of the provenance__ (variable `gg`) and a __random block effect__ (variable `bl`). 2. Specify sensible __initial variances__ for the random components 3. Extend `fm1` by including an __interaction__ between cross-classified variables --- # Random or fixed? - Can I change variables from __fixed__ to __random__ and viceversa? - How that would change the results and their __interpretation__? --- # Initial variances - Good practice: specify __initial values__ for all the variances in the model - `breedR` provides sensible default values, but sometimes you can help ```r fm1 <- remlf90( ···, * var.ini = list(···, resid = ···) ) ``` --- # Interactions - What is an __interaction effect__? - When can we estimate interactions? --- # Nested or crossed variables ## A property of the __data__, not the model - In globulus, the **family** (`mum`) is _nested_ within the **provenance** (`gg`) - This is a matter of (good) codification: .pull-left[ Nested factors |gg | mum | |---:|:----| |A | 1 | |A | 2 | |B | 3 | |B | 4 | ] .pull-right[ Cross-classified factors |gg | mum | |---:|:----| |A | 1 | |A | 2 | |B | 1 | |B | 2 | ] --- # Specifying interactions in breedR ## Create a new variable!! - See `?base::interaction` - If F1 and F2 are factors ```r F12 <- factor(F1:F2) ``` is another factor with all the __observed__ level combinations - Use this variable as another __term__ in the model --- class: middle, inverse # Excercise 3. ## Extract results 1. Use functions `summary()`, `fixef()` and `ranef()` to extract __estimates__ 2. __Variance estimates__ are stored in `fm2$var` 3. __Plot__ observed phenotype and `residual()`s vs `fitted()` values to assess the model predictive ability and to diagnose residuals --- class: middle, inverse # Excersise 4. ## Pedigrees 1. Replace the genetic variable `mum` at _family_ level, by an _animal model_ with a genetic effect at _individual_ level. 2. Retrieve the predicted individual breeding values. Make sure they are in the same order as observed in the globulus dataset. --- # The _animal_ model $$ y \sim \mu_0 + a + \varepsilon$$ with `\(a \sim \mathcal N(0, \sigma_a^2 \mathrm{A})\)` and `\(\varepsilon \sim \mathcal N(0, \sigma^2)\)` where `\(\mathrm{A}\)` is the __relationship matrix__, derived from the __pedigree__ .center[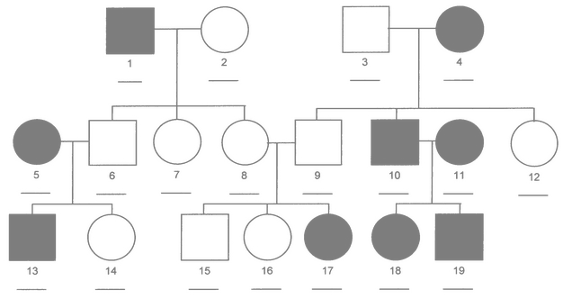] --- # Additive Genetic Effect - Random effect at **individual level** - Based on a **pedigree** - BLUP of **Breeding Values** from own and relatives' phenotypes - Represents the **additive component** of the genetic value - More __general__: - family effect is a particular case - accounts for more than one generation - mixed relationships - More __flexible__: allows to select individuals within families --- # Specification in `breedR` ```r fm3 <- remlf90( fixed = ···, random = ···, * genetic = list( * model = 'add_animal', # model name for the term * pedigree = ···, # pedigree (see Details in ?remlf90) * id = ···), # variable name of the individual data = globulus ) ``` --- # Specifying a _pedigree_ - A 3-column `data.frame` or `matrix` with the codes for each individual and its parents - A **family** effect is easily translated into a pedigree: - use the **family code** as the identification of a fictitious **mother** - use `0` or `NA` as codes for the **unknown fathers** <table> <thead> <tr> <th style="text-align:right;"> self </th> <th style="text-align:right;"> dad </th> <th style="text-align:right;"> mum </th> </tr> </thead> <tbody> <tr> <td style="text-align:right;"> 69 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 64 </td> </tr> <tr> <td style="text-align:right;"> 70 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 41 </td> </tr> <tr> <td style="text-align:right;"> 71 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 56 </td> </tr> <tr> <td style="text-align:right;"> 72 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 55 </td> </tr> <tr> <td style="text-align:right;"> 73 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 22 </td> </tr> <tr> <td style="text-align:right;"> 74 </td> <td style="text-align:right;"> 0 </td> <td style="text-align:right;"> 50 </td> </tr> </tbody> </table> --- # Retrieving BVs from the fitted model ## Be careful about the __ordering__ - `ranef(·)$genetic` typically __does not match__ the order of observed individuals (founders, clones, recoding) - `\(Z\,a\)` = `model.matrix(·)$genetic %*% ranef(·)$genetic` always give the PBV for all observed individuals in the dataset, in that order. --- # Sorting BVs .pull-left[ Observed data: <table> <thead> <tr> <th style="text-align:right;"> self </th> <th style="text-align:right;"> dad </th> <th style="text-align:right;"> mum </th> </tr> </thead> <tbody> <tr> <td style="text-align:right;"> 4 </td> <td style="text-align:right;"> 1 </td> <td style="text-align:right;"> 2 </td> </tr> <tr> <td style="text-align:right;"> 3 </td> <td style="text-align:right;"> 1 </td> <td style="text-align:right;"> 2 </td> </tr> </tbody> </table> `$$\underbrace{ \begin{bmatrix} 0 & 0 & 0 & 1 \\ 0 & 0 & 1 & 0 \end{bmatrix}}_\text{model.matrix(·)\$genetic} \cdot \underbrace{ \begin{bmatrix} a_1 \\ a_2 \\ a_3 \\ a_4 \end{bmatrix}}_\text{ranef(·)\$genetic} = \begin{bmatrix} a_4\\ a_3 \end{bmatrix}$$` ] .pull-right[ Implied pedigree: <table> <thead> <tr> <th style="text-align:right;"> self </th> <th style="text-align:right;"> dad </th> <th style="text-align:right;"> mum </th> </tr> </thead> <tbody> <tr> <td style="text-align:right;"> 1 </td> <td style="text-align:right;"> NA </td> <td style="text-align:right;"> NA </td> </tr> <tr> <td style="text-align:right;"> 2 </td> <td style="text-align:right;"> NA </td> <td style="text-align:right;"> NA </td> </tr> <tr> <td style="text-align:right;"> 3 </td> <td style="text-align:right;"> 1 </td> <td style="text-align:right;"> 2 </td> </tr> <tr> <td style="text-align:right;"> 4 </td> <td style="text-align:right;"> 1 </td> <td style="text-align:right;"> 2 </td> </tr> </tbody> </table> 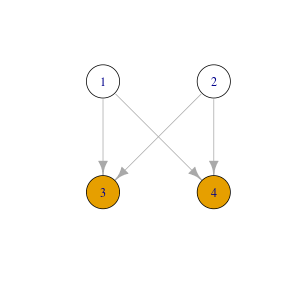<!-- --> ] --- class: middle, inverse # Excercise 5. ## Heritability Compute the heritability of the globulus dataset using the three available methods. --- # Methods for computing heritability |Method | Limitations | |:-------|:------------| |1. Automatic | `genetic` term<br> method `ai`<br> fixed formula | |2. Explicit Formula | method `ai`<br> | |3. Bootstrap | - <br> | See: `RShowDoc("Heritability", package = "breedR")`. --- # Formula syntax For a single trait model such as: ```r fm <- remlf90( fixed = y ~ x1 + x2, random = u + v, genetic = ···, * progsf90.options = paste('se_covar_function h2', h2fml), data = globulus ``` count the model terms in order as: | term: | x1 | x2 | u | v | `genetic` | |------:|:--|:--|:--|:--|:--| | i: | 1 | 2 | 3 | 4 | 5 | And build a formula string __without spaces__ as in: ```r h2fml <- 'G_5_5_1_1/(G_5_5_1_1+G_4_4_1_1+R_1_1)' ``` --- # Bootstrap procedure 1. __Fit__ the model to your data. 2. Write a function to __simulate observations__ from the fitted model. You can use `breedR.sample.phenotype()` if the model is simple enough ```r resample_data <- function(fit) { ## Use the estimated values in fit to produce a new data.frame ## of the same size and with the same variables as globulus. return(dat) } ``` 3: write a function to fit a simulated dataset and extract the target values ```r sim_target() <- function(dat) { ## Fit the same model as fm3 to this fake dataset dat ## Return the point estimates of all variances and heritability return(estimates) } ``` --- # Resample parameters of interest 4: Replicate the data-generation+estimation process many times __Warning__: this can take a while, depending on the model and the dataset. ```r boot_estimates <- function(N, fit) { ans <- replicate(N, sim_target(dat = resample_globulus(fit))) return(as.data.frame(t(ans))) } empirical_dist <- boot_estimates(N = 100, fit = fm3) ``` --- class: inverse, center, middle background-image: url(img/breedRhex.png) background-position: 50% 80% ## This should account for most analyses of 'simple' progeny tests